My lab studies the epigenetic mechanisms of brain cancer and neurodevelopmental syndromes, with the goal of understanding how diseases are initiated and maintained. We utilize an integrative approach that leverages multiple experimental methods, including patient-derived samples, cerebral organoids, and transgenic animals, along with state-of-the-art sequencing technologies to profile the genome, epigenome, and transcriptome.
Current research projects
- Understanding how histone modifier mutations alter cell fate in the brain
- Identifying epigenetic drivers of childhood brain tumors
- Examining epigenetic rewiring as a therapeutic resistance mechanism
Epigenetic regulation of development and disease
DNA and histones are covalently modified to control transcriptional activity, this dynamic “epi-genetic” regulation is critical for normal development. Indeed, mutations in histones or histone modifying enzymes can lead to defects during cellular differentiation and contribute to neoplastic transformation.
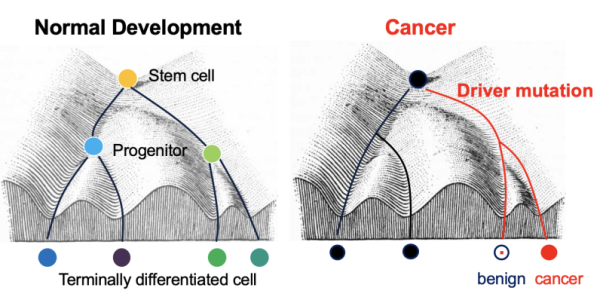
The epigenetic landscape determines developmental & oncogenic potential. Conrad Waddington famously described a metaphor for developmental epigenetics - imagine a stem cell as a marble rolling downhill, being restricted in certain cell fates by the grooves and valleys of the underlying chromatin landscape. In cancer, the same epigenetic landscape may also restrict capacity for oncogenic transformation in a given cell-of-origin.
Childhood brain cancer as epigenetic disorders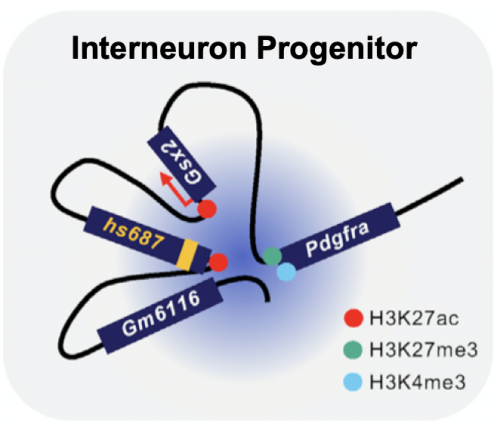
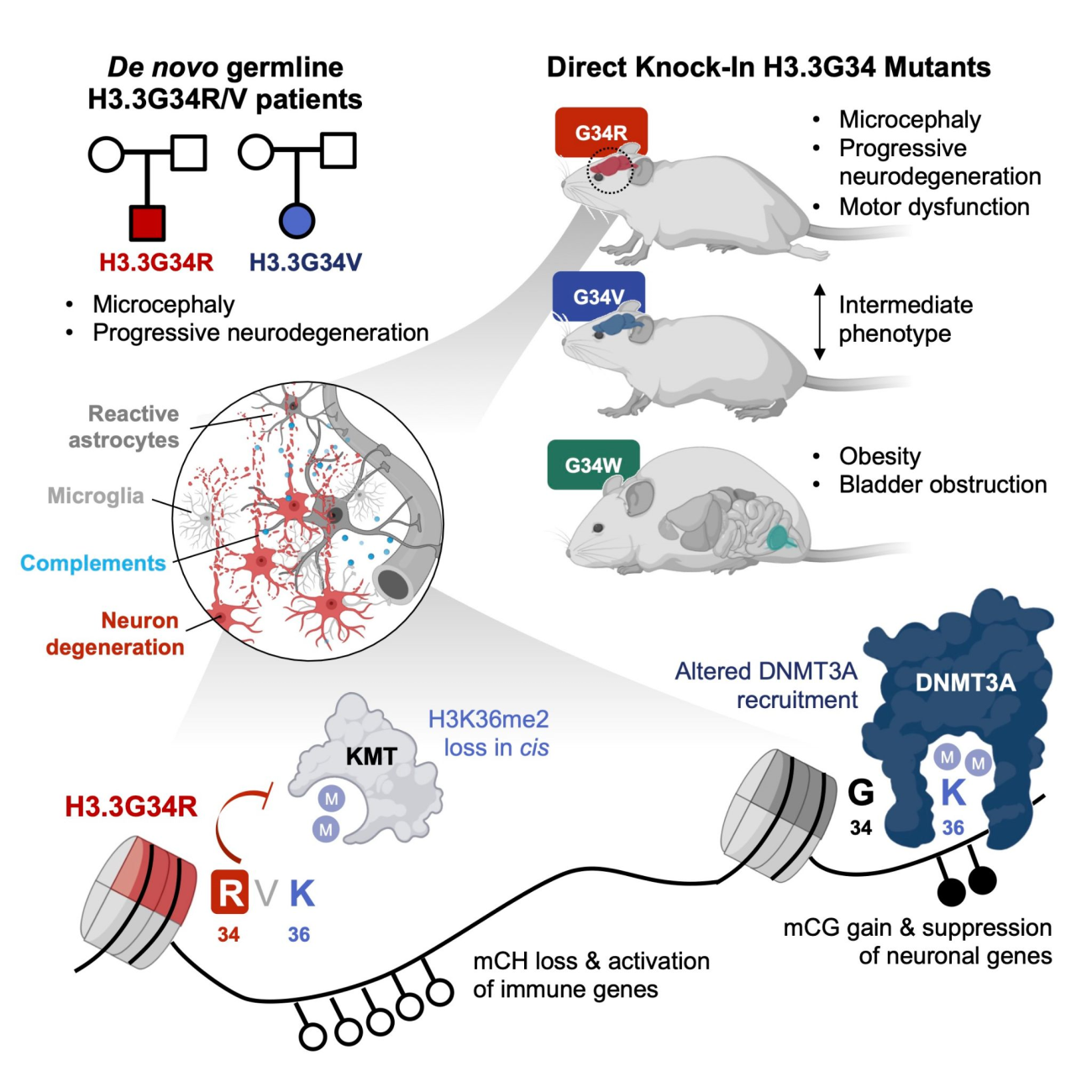
High-grade glioma (HGG) is a lethal form of primary brain cancer and the leading cause of mortality in children and young adults. These tumors frequently bear hotspot mutations in genes encoding histone H3 or histone-modifying enzymes, and many are considered epigenetic disorders. In a previous study, we demonstrated that despite being classified as gliomas, H3.3G34R/V brain tumors actually originate from a committed progenitor of the interneuron lineage (Chen et al., 2020). Within this progenitor, there is a developmentally regulated chromatin conformation that positions the oncogene PDGFRA in close proximity to an activated enhancer (hs687). We hypothesize that this unique chromatin context in the cell-of-origin facilitates oncogenic transformation by inappropriately activating PDGFRA. PDGFRA mutations are present in 40% of H3.3G34R/V HGGs at diagnosis, which further increases to 80% at recurrence. Clinically, PDGFRA encodes a receptor tyrosine kinase that can be targeted with small molecule inhibitors. Therefore, our findings have real implications for the treatment of H3.3G34R/V HGGs.
Germline histone mutations in neurodevelopmental syndromes
Recently, 63 histone H3.3 mutations were found in the germline of patients with neurodevelopmental disorders. While most of the germline H3.3 mutations do not overlap with oncohistone mutations, the H3.3G34R and G34V mutations were found in patients who exhibit global developmental delay, microcephaly, and progressive neurodegeneration. Using an endogenous knock-in transgenic murine model, we showed that the G34R, G34V, and G34W mutants have drastically different phenotypes (Khazaei et al., 2023). Specifically, G34R causes fully penetrant neurological deficits, microcephaly, and lethal ataxia. Molecularly, the G34R mutation hinders deposition of an epigenetic modification (CH methylation) that functions uniquely in mature neurons. As a result of this epigenetic disruption, G34R neurons repress gene expression programs for synaptogenesis, instead aberrantly activating immunity genes to induce a persistent state of inflammation. Our study sheds molecular insights on how histone H3.3G34R mutations cause disease specifically in the developing brain.