Scientific Highlights
2. Discovered tetrameric antibody complexes, reagents that are key to the success of Vancouver's STEMCell Technologies.
In Amsterdam, I made mouse IgG1 monoclonal antibodies to peroxidase to link peroxidase to cell surface antigens via mouse IgG1 monoclonal antibodies against cell surface antigens using polyclonal anti-mouse IgG1 (1,2). Based on the bivalency of IgG antibodies and the presence of two identical heavy chains in each of such antibody molecules, I speculated that monoclonal antibodies specific for selected epitopes present on the heavy chain of mouse IgG1 molecules might be able to cross-link two mouse IgG1 monoclonal antibodies into a stable tetrameric antibody complex. To test this hypothesis, I made (and found!) suitable (rat) monoclonal antibodies specific for mouse IgG1 (3). See Figure 1.
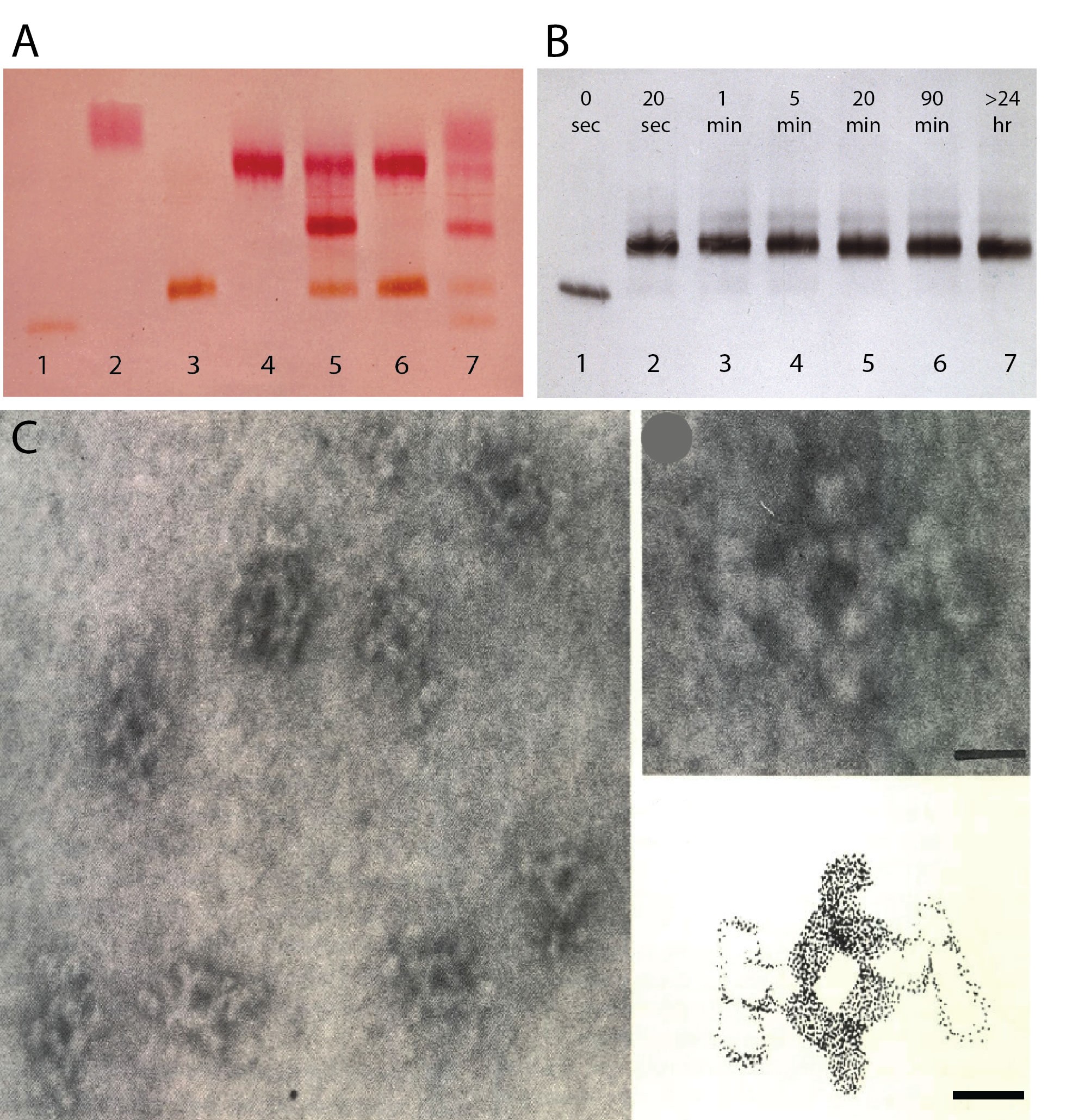
Figure 1. Tetrameric complexes of monoclonal rat and mouse antibodies (from (3). A. Staining for peroxidase and alkaline phosphatase following agar gel electrophoresis of mouse IgG1 anti-enzyme antibodies before and after addition of selected monoclonal rat anti-mouse IgG1. Lane 1: mouse IgG1 anti-peroxidase (a). Lane 2: mouse IgG1 anti- alkaline phosphatase (b). Lane 3: mouse IgG1 anti peroxidase plus an equimolar amount of rat anti-mouse IgG1. Note: no free mouse antibody. Lane 4: mouse IgG1 anti- alkaline phosphatase plus rat anti-mouse IgG1. Lane 5: equimolar amounts of mouse IgG1 anti- peroxidase and mouse IgG1 anti- alkaline phosphatase mixed before addition of an equimolar amount of rat anti-mouse IgG1. Note middle band with bispecific monoclonal tetrameric antibody complexes! Lane 6: A mixture of the complexes shown in lane 3 and 4. Note the absence of bispecific complexes, indicating high stability of tetrameric antibody complexes. Lane 7. As lane 5, using a limiting amount of rat anti-mouse IgG1 which shows free mouse IgG1 antibodies. B. Time course of tetrameric antibody complex formation. Mouse IgG1 anti-peroxidase (lane 1) was mixed with an equimolar amount of monoclonal rat anti-mouse IgG1. At the indicated time points the mixture was subjected to agarose gel electrophoresis and anti-peroxidase was visualized after blotting as in A. Note that antibody complexes are formed within seconds and not affected by electrophoresis. C. Scanning electron microscopy of purified tetramolecular antibody complexes. Scale bar 10 nm.
The ability to easily crosslink two different monoclonal antibodies into stable tetrameric antibody complexes (US patent No. 4,868,109) has found numerous applications in biotechnology. For example, we described applications in cell separation (4,5) and fluorescent labeling of cell surface antigens (6). Tetrameric antibody complexes are key to cell separation and many other technologies commercialized by STEMCELL Technologies in Vancouver, the largest biotechnology company in Canada.
References:
- Lansdorp, P.M., Astaldi, G.C., Oosterhof, F., Janssen, M.C. and Zeijlemaker, W.P. (1980) Immunoperoxidase procedures to detect monoclonal antibodies against cell surface antigens. Quantitation of binding and staining of individual cells. J Immunol Methods, 39, 393-405.
- Lansdorp, P.M., van der Kwast, T.H., de Boer, M. and Zeijlemaker, W.P. (1984) Stepwise amplified immunoperoxidase (PAP) staining. I. Cellular morphology in relation to membrane markers. J Histochem Cytochem, 32, 172-178.
- Lansdorp, P.M., Aalberse, R.C., Bos, R., Schutter, W.G. and Van Bruggen, E.F. (1986) Cyclic tetramolecular complexes of monoclonal antibodies: a new type of cross-linking reagent. Eur J Immunol, 16, 679-683.
- Thomas, T.E., Abraham, S.J., Otter, A.J., Blackmore, E.W. and Lansdorp, P.M. (1992) High gradient magnetic separation of cells on the basis of expression levels of cell surface antigens. J Immunol Methods, 154, 245-252.
- Thomas, T.E., Sutherland, H.J. and Lansdorp, P.M. (1989) Specific binding and release of cells from beads using cleavable tetrameric antibody complexes. J Immunol Methods, 120, 221-231.
- Wognum, A.W., Thomas, T.E. and Lansdorp, P.M. (1987) Use of tetrameric antibody complexes to stain cells for flow cytometry. Cytometry, 8, 366-371.
8. Reported that without telomerase mice lose around 5kb of telomeric DNA per generation.
With PNA and Q-FISH tools, we contributed to a number of important studies, the most cited one being our contribution to the study of the telomerase knock-out (KO) KO mouse (1) (> 2000 citations). This paper was already under review at Cell when we got involved. Our findings significantly changed the message, essentially from “telomerase is not as important as we believed: it can be knocked out without consequences” to “telomerase KO mice lose 5 kb of telomeric DNA with each generation and uncapped telomeres at late generations cause chromosome fusions and genome instability”. See Figure.
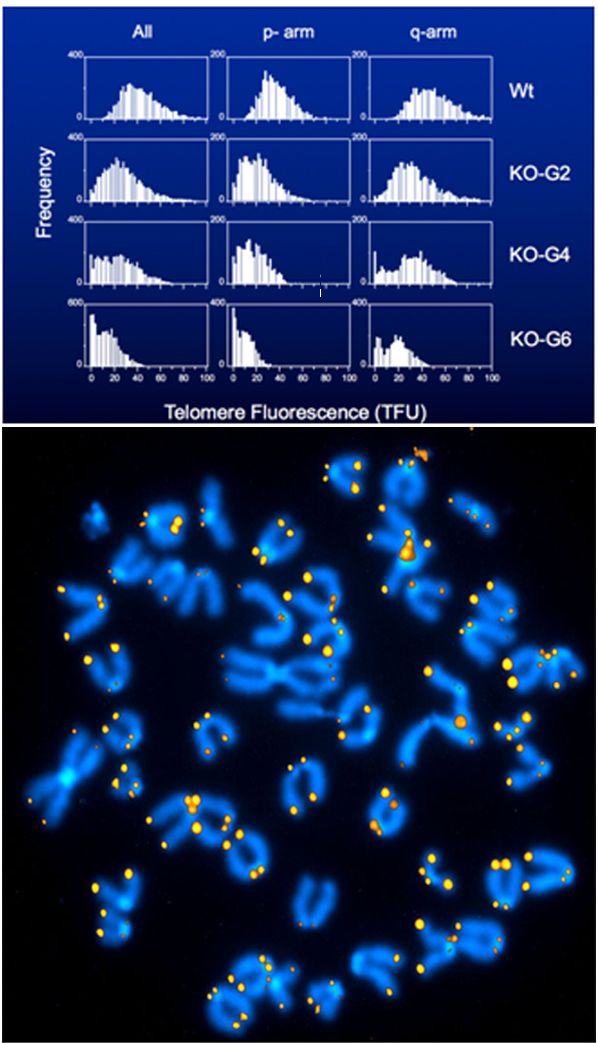
Figure. Telomerase KO mice lose about 5 kb of telomere repeats per generation. Q-FISH analysis of embryonic fibroblasts from the indicated subsequent generations of telomerase KO mice. 1 TFU corresponds to ~1kb of telomere repeats. Note that in wild-type (WT) animals all chromosomes are “capped” with readily detectable telomere repeats, whereas at generation 6 (bottom panel and chromosome spread) many chromosome ends have no telomere repeats. Note the dicentric chromosomes without telomere repeats at the point of fusion point (e.g. at 9 pm).
Reference:
- Blasco, M.A., Lee, H.W., Hande, M.P., Samper, E., Lansdorp, P.M., DePinho, R.A. and Greider, C.W. (1997) Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell, 91, 25-34.
12. Founded Repeat Diagnostics, a company that provides clinical measurements of the average length of telomere repeats at chromosome ends in nucleated blood cells.
We developed and perfected a method to measure the average length of telomeres in specific nucleated cell types in peripheral blood using PNA probes, fluorescence in situ hybridization (FISH) and flow cytometry, a technique we called flow FISH (1). With this method, we showed that there is a large variation in the average telomere length between individuals of the same age and that the most dramatic decline in telomere length occurs in the first few years of life (see Figure). Interestingly, at birth and throughout life the average telomere length was found to be longer in females than in males (2) and Figure, left panel). Based on reported sex differences in gene expression between cells from human embryos, I recently proposed that such telomere length differences could be established before embryo implantation (3). In collaboration with hematologists and geneticists at NIH, we established that the telomere length in patients with genetic defects in telomere pathway genes, such as TERT, TERC, RTEL1 and others could readily be distinguished from patients with other causes for their bone marrow failure (reviewed in (4). Because our flow FISH measurements could also distinguish carriers from telomerase mutations from siblings without such genetic defect (Figure, right panel), we obtained many requests from clinicians to perform diagnostic flow FISH experiments for their patients in our research lab. This clearly was not ideal, and I decided to set up a company, Repeat Diagnostics Inc., to provide clinical telomere length measurements. This company employs 8 people and is processing 10-20 samples per day for clinicians around the world looking after patients suspected of inherited or acquired “telomere biology disorders”. My take on the role of telomeres and telomerase in aging and cancer is summarized in two review papers (5,6).
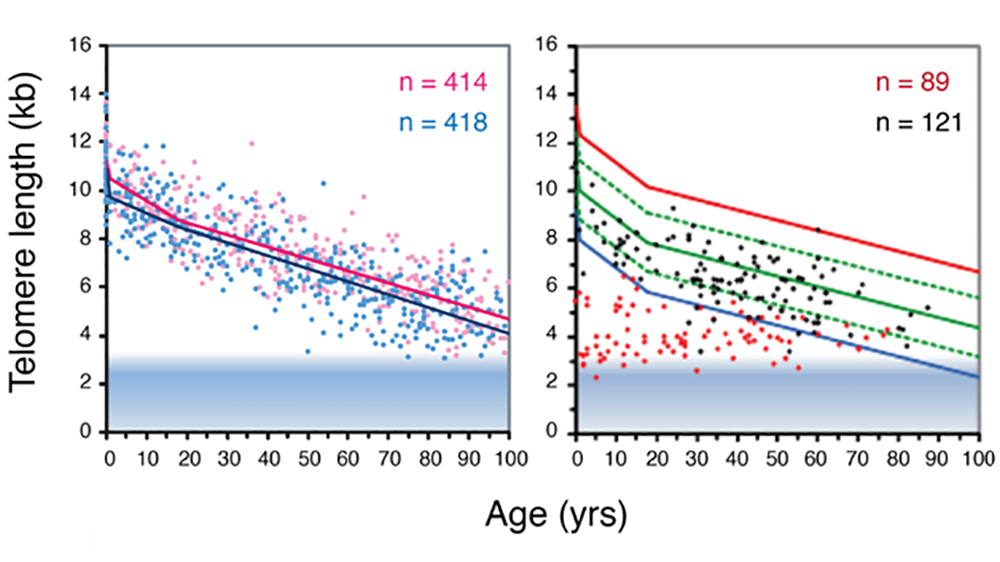
Figure. Telomere length decline with age in human lymphocytes in peripheral blood measured using flow FISH. The left panel shows that a difference in average telomere length between females (pink) and males (blue) persist throughout life. The right panel shows the telomere length in heterozygous carriers of a mutation in either the TERC or the TERT gene (red) and their unaffected siblings (black). Blue line: bottom 1 percent of normal telomere length at indicated age. Figure adapted from (2).
References:
- Baerlocher, G.M., Vulto, I., de Jong, G. and Lansdorp, P.M. (2006) Flow cytometry and FISH to measure the average length of telomeres (flow FISH). Nat Protoc, 1, 2365-2376.
- Aubert, G., Baerlocher, G.M., Vulto, I., Poon, S.S. and Lansdorp, P.M. (2012) Collapse of telomere homeostasis in hematopoietic cells caused by heterozygous mutations in telomerase genes. PLoS Genet, 8, e1002696.
- Lansdorp, P.M. (2022) Sex differences in telomere length, lifespan, and embryonic dyskerin levels. Aging Cell, 21, e13614.
- Savage, S.A. (2025) Telomeres and Human Disease. Cold Spring Harb Perspect Biol, 17.
- Lansdorp, P.M. (2022) Telomeres, aging, and cancer: the big picture. Blood, 139, 813-821.
- Lansdorp, P.M. (2022) Telomeres, Telomerase and Cancer. Arch Med Res, 53, 741-746.
15. Developed the single cell Strand-seq method.
Encouraged by the results described in our Nature paper (1), we embarked on developing the single cell DNA template strand sequencing (Strand-seq) technique (2). At the time, we could not have predicted the wide range of applications that this technique would have outside our initial goal: to distinguish sister chromatids in order to test the “silent sister” hypothesis. Strand-seq results are illustrated in the Figure and in a recent review (3).
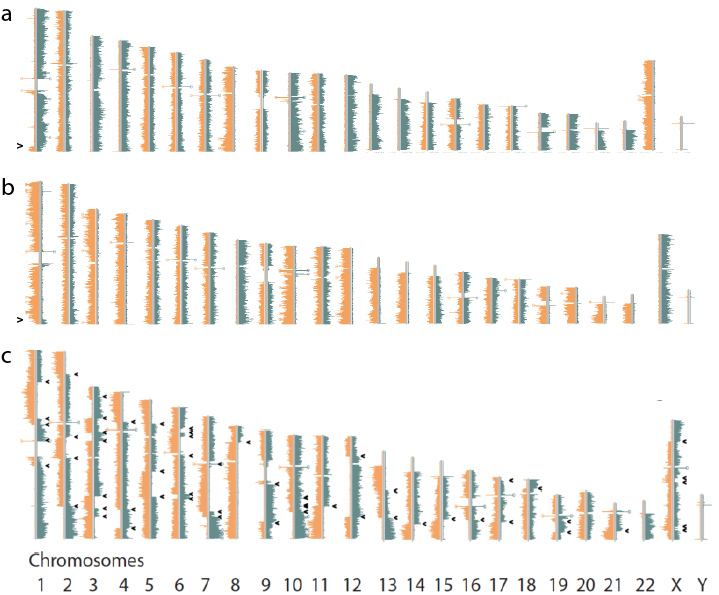
Figure. Strand-seq allows identification and analysis of sister chromatids. The two daughter cells (a and b) of a single parental CD34+ cell show perfect, complementary segregation of parental DNA template strands and only a single sister chromatid exchange event (arrow, bottom chr 1). Note that all chromosomes showing reads mapping to both strands of the reference genome (Chr 2, 5, 6, 7, 9, 11, 14, 16, 17 and 18 in this cell pair) can be used to map SNPs to parental genomes along entire chromosomes and assemble complete physical maps of parental haplotypes. c) Sister chromatid exchange events are very common in cells from patients with Bloom’s syndrome. In this cell 56 SCE events are detected (arrows).
References:
- Falconer, E., Chavez, E.A., Henderson, A., Poon, S.S., McKinney, S., Brown, L., Huntsman, D.G. and Lansdorp, P.M. (2010) Identification of sister chromatids by DNA template strand sequences. Nature, 463, 93-97.
- Falconer, E., Hills, M., Naumann, U., Poon, S.S., Chavez, E.A., Sanders, A.D., Zhao, Y., Hirst, M. and Lansdorp, P.M. (2012) DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution. Nat Methods, 9, 1107-1112.
- Hanlon, V.C.T. and Lansdorp, P.M. (2026) Strand-seq and the future of personalized genomics. Nat Genet.
16. Generated comprehensive maps of human polymorphic inversions.
My lab generated comprehensive maps of polymorphic inversions in the human genome. This work was spearheaded by three graduate students: Ashley Sanders, David Porubsky and Vincent Hanlon. The Figure shows examples of results. Following Ashley’s initial papers (1,2), David reported on inversions and genome instability (3) and Vincent developed software to visualize phased inversions in a genome browser (4).
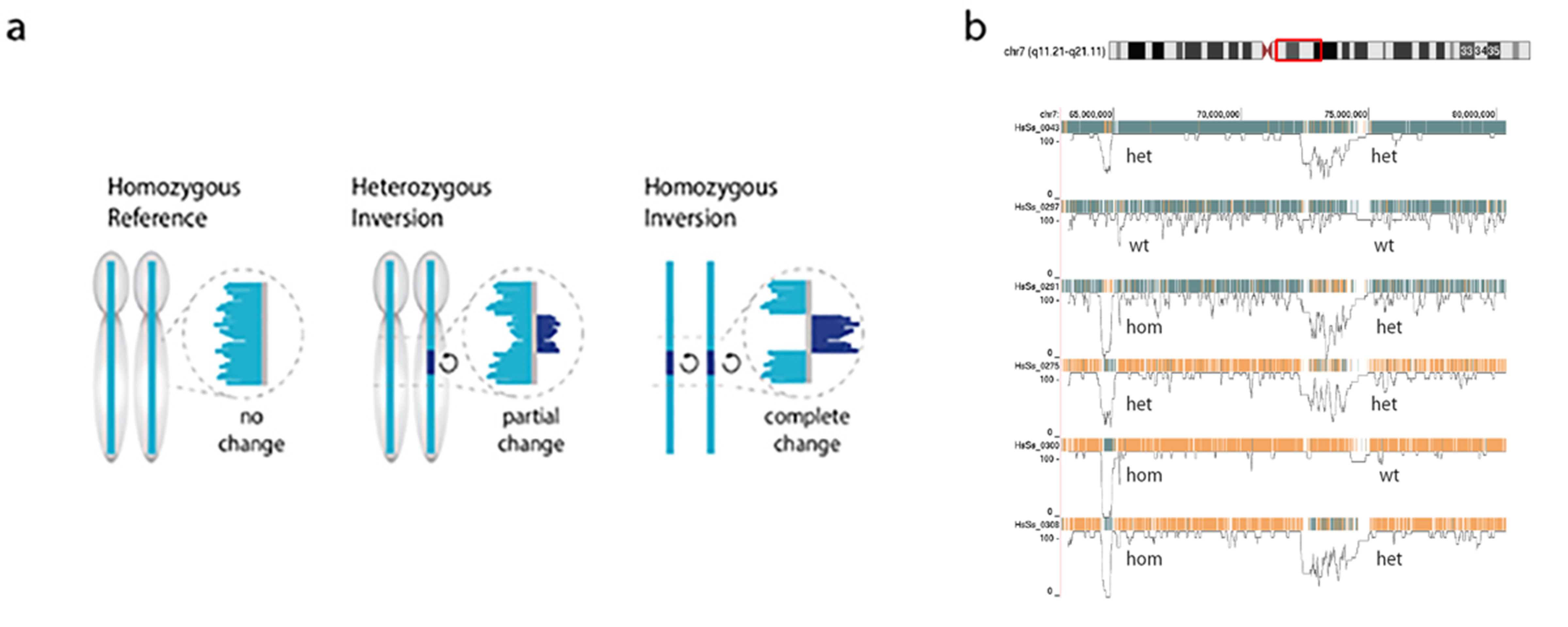
Figure. Sequence reads generated by Strand-seq map either concordant with the human reference genome sequence (a, left) or map to both strands of the reference genome (reflecting a heterozygous inversion (a, middle panel) or completely to the opposite strand of the reference genome reflecting a homozygous inversion (a, right panel). Polymorphic inversions in the human genome generate a large amount of genetic diversity between individuals as is shown in b for a 20Mb segment of chr 7 (red box) for 6 individuals. Note that the role of this type of genetic diversity is largely unexplored because of the difficulty to generate genome-wide information about polymorphic inversions using other sequence modalities.
References:
- Sanders, A.D., Hills, M., Porubsky, D., Guryev, V., Falconer, E. and Lansdorp, P.M. (2016) Characterizing polymorphic inversions in human genomes by single-cell sequencing. Genome Res, 26, 1575-1587.
- Chaisson, M.J.P., Sanders, A.D., Zhao, X., Malhotra, A., Porubsky, D., Rausch, T., Gardner, E.J., Rodriguez, O.L., Guo, L., Collins, R.L. et al. (2019) Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat Commun, 10, 1784.
- Porubsky, D., Hops, W., Ashraf, H., Hsieh, P., Rodriguez-Martin, B., Yilmaz, F., Ebler, J., Hallast, P., Maria Maggiolini, F.A., Harvey, W.T. et al. (2022) Recurrent inversion polymorphisms in humans associate with genetic instability and genomic disorders. Cell, 185, 1986-2005 e1926.
- Hanlon, V.C.T., Mattsson, C.A., Spierings, D.C.J., Guryev, V. and Lansdorp, P.M. (2021) InvertypeR: Bayesian inversion genotyping with Strand-seq data. BMC Genomics, 22, 582.
17. Developed novel ways to establish haplotypes along entire chromosomes.
The diploid nature of the genome is often incompletely represented in “whole genome sequence” analysis, where a genome is typically represented as a set of unphased variants with respect to a reference genome. However, important biological phenomena such as compound heterozygosity and epistatic effects between enhancers and target genes can only be studied when haplotype-resolved genomes are available. Hence a method that can produce dense and accurate chromosome-length haplotypes is highly desirable. Using a well-studied trio from the HapMap consortium we showed that Strand-seq allows for accurate phasing along entire chromosomes (1). The principle of this method and phasing trio family members are shown in Figure 1.
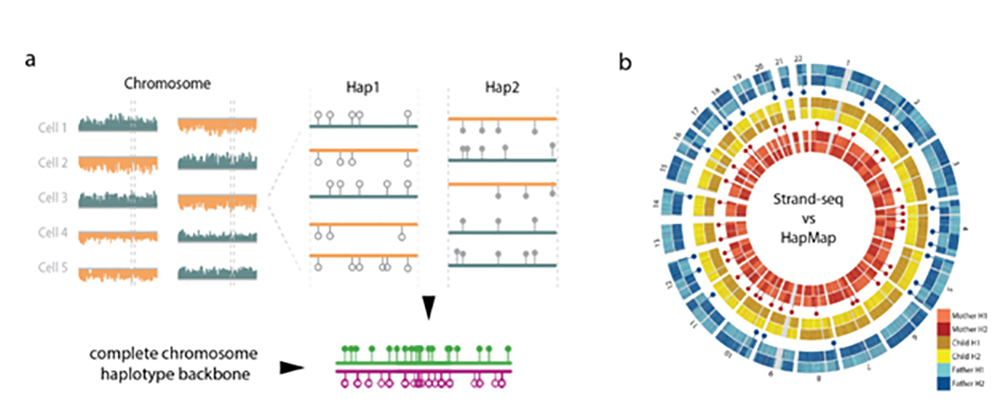
Figure 1. a) Strand-seq provides a physical haplotype map of SNP’s along entire chromosomes. Multiple cells with Strand-seq sequence reads mapping to both strands of the reference genome for a given chromosome (orange and blue) are used to assemble haplotype backbones (1). Such chromosome-long haplotypes correspond accurately (>99%) with HapMap reference data for a child (b, yellow) but not her parents (father blue , mother red), where Strand-seq reveals the location of parental meiotic recombination events (resp. red and blue dots). The haplotype backbones generated by Strand-seq allow complete phasing of human genomes when combined with other type of sequence data (e.g. 10X or PacBio data (1, 2, 3).
When combined with other type of sequencing data, Strand-seq allows near complete phasing (>99% of SNP’s) of human genomes (Fig 2, from 2).
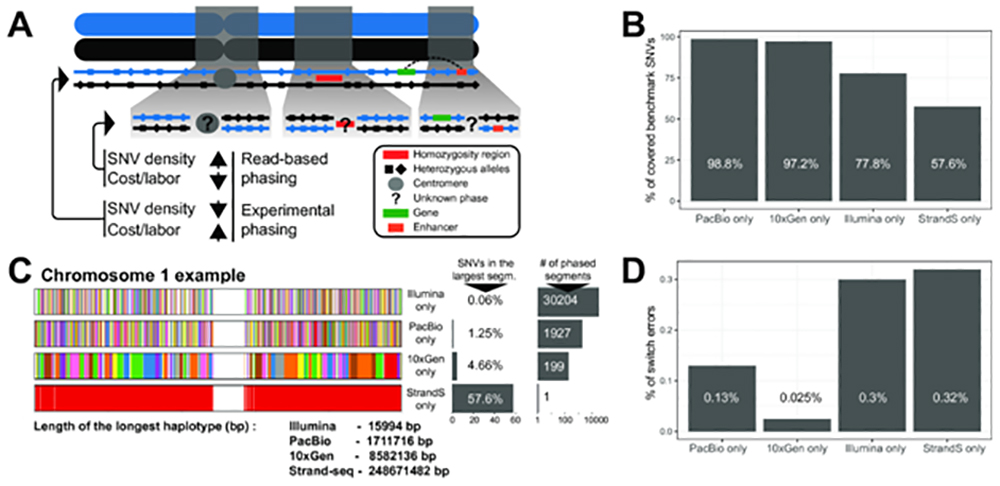
Figure 2. A) Two homologous chromosomes are shown (blue and black). Experimental phasing approaches like Strand-seq can connect heterozygous alleles along whole chromosomes, however, at higher costs (time and labor) and lower density of captured alleles. In contrast, read-based phasing can deliver high-density haplotypes, but only short haplotype segments are assembled with an unknown phase between them. B) Barplot showing the percentage of phased variants, for each sequencing technology, from the total number of reference SNVs (Illumina platinum haplotypes). C) Graphical summary of phased haplotype segments for Illumina, PacBio, 10X Genomics and Strand-seq phasing shown for chromosome 1. Each haplotype segment is colored in a different color with the longest haplotype colored in red. Side bargraph reports the percentage of SNVs phased in the longest haplotype segment. D) Accuracy of each independent phasing approach measured as percentage short switch errors in comparison to benchmark haplotypes.
References:
- Porubsky, D., Sanders, A.D., van Wietmarschen, N., Falconer, E., Hills, M., Spierings, D.C., Bevova, M.R., Guryev, V. and Lansdorp, P.M. (2016) Direct chromosome-length haplotyping by single-cell sequencing. Genome Res, 26, 1565-1574.
- Porubsky, D., Garg, S., Sanders, A.D., Korbel, J.O., Guryev, V., Lansdorp, P.M. and Marschall, T. (2017) Dense and accurate whole-chromosome haplotyping of individual genomes. Nat Commun, 8, 1293.
- Hanlon, V.C.T. and Lansdorp, P.M. (2026) Strand-seq and the future of personalized genomics. Nat Genet.
18. Improved Strand-seq library preparation protocols.
Although Strand-seq data are increasingly in demand, the preparation of single cell Strand-seq libraries is technically challenging (1). Variable results and limited informative reads have hampered widespread application of the Strand-seq method despite its theoretical appeal. We recently reported a simplified “one pot” (OP) method to make Strand-seq libraries that overcomes many of the previous technical challenges (2). Specifically, we developed a nanoliter-volume protocol in which reagents are added cumulatively, DNA purification steps are avoided, and enzymes are inactivated with a thermolabile protease. OP-Strand-seq libraries routinely capture 10%–25% of the genome from a single cell with reduced costs and increased throughput, opening up a wide variety of studies that are being pursued in collaboration with investigators in and outside Vancouver and Canada. A typical result of our current Strand-seq results is shown in the Figure and various issues related to Strand-seq library production are reviewed in (3).
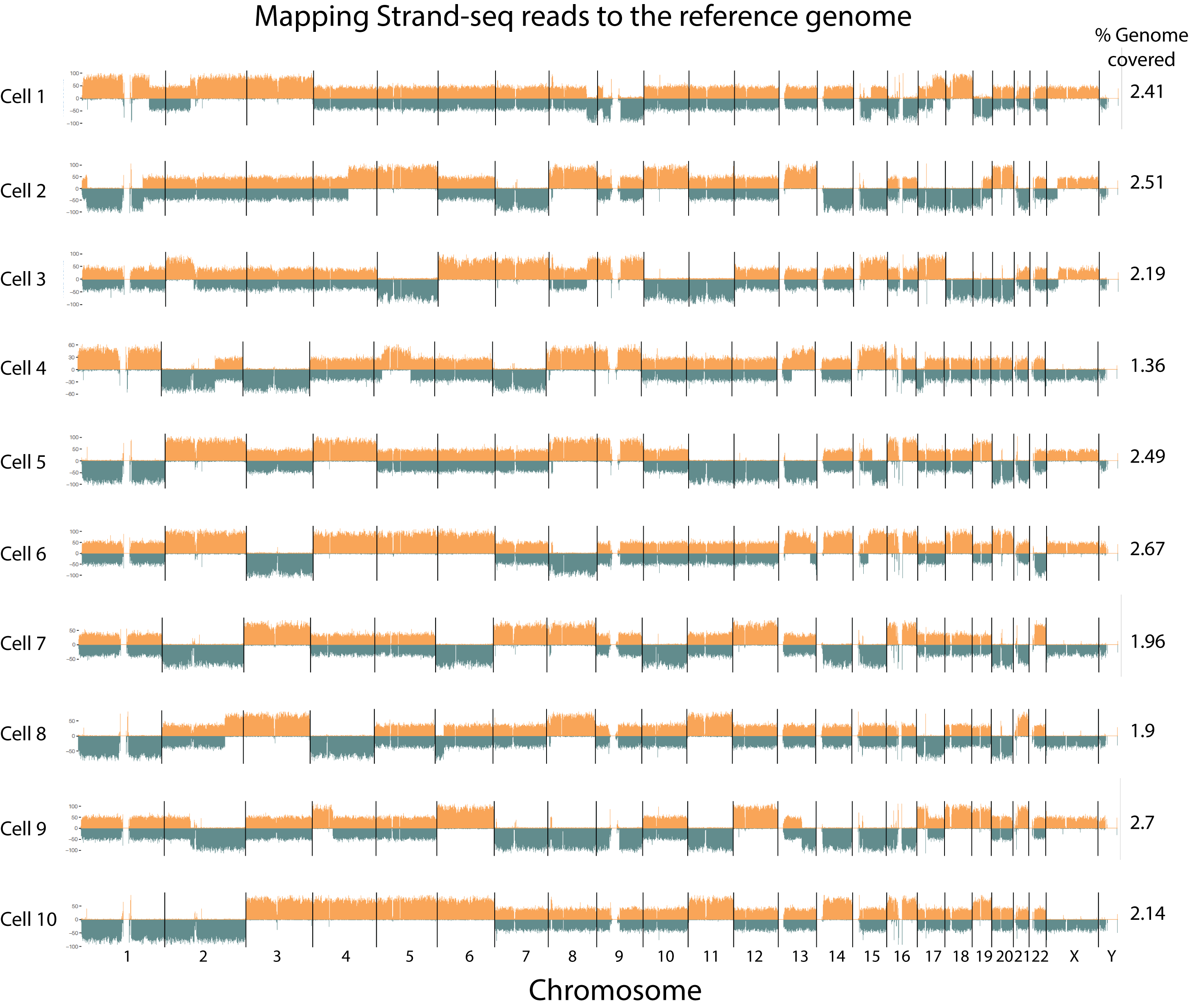
Figure. Example of Strand-seq results (data from 4). Reads mapping to either the plus (+) strand of the human reference genome HG38 (“Crick” Orange) or the minus (-) strand of HG38 (“Watson”, blue) are shown for all chromosomes in single cells from this male (X/Y) individual. Available libraries were not sequenced deeply and the percentage of a haploid genome captured is shown on the right.
References:
- Sanders, A.D., Falconer, E., Hills, M., Spierings, D.C.J. and Lansdorp, P.M. (2017) Single-cell template strand sequencing by Strand-seq enables the characterization of individual homologs. Nat Protoc, 12, 1151-1176.
- Hanlon, V.C.T., Chan, D.D., Hamadeh, Z., Wang, Y., Mattsson, C.A., Spierings, D.C.J., Coope, R.J.N. and Lansdorp, P.M. (2022) Construction of Strand-seq libraries in open nanoliter arrays. Cell Rep Methods, 2, 100150.
- Hanlon, V.C.T. and Lansdorp, P.M. (2026) Strand-seq and the future of personalized genomics. Nat Genet.
- Porubsky, D., Dashnow, H., Sasani, T.A., Logsdon, G.A., Hallast, P., Noyes, M.D., Kronenberg, Z.N., Mokveld, T., Koundinya, N., Nolan, C. et al. (2025) Human de novo mutation rates from a four-generation pedigree reference. Nature, 643, 427-436.
19. Assigning alleles to parent-of-origin chromosomes.
Genetic testing can inform a patient’s inherited risk for developing a disease. However, predicting which side of the family an autosomal variant comes from is often a limitation of current technology. Inability to determine a variant’s parent-of-origin (PofO) (i.e., if it is maternally or paternally inherited) can lead to uncertain patient management and ineffective genetic testing of family members. In collaboration with Intan Schrader, Steven Jones and their collaborators we have developed a protocol using Oxford Nanopore Technologies’ long read sequencing (ONT-LRS) in combination with chromosome-length haplotyping enabled by single cell Strand-seq to allow, for the first time, PofO haplotyping without any sequence information of the parents (1). The principle of this method is shown in the Figure.
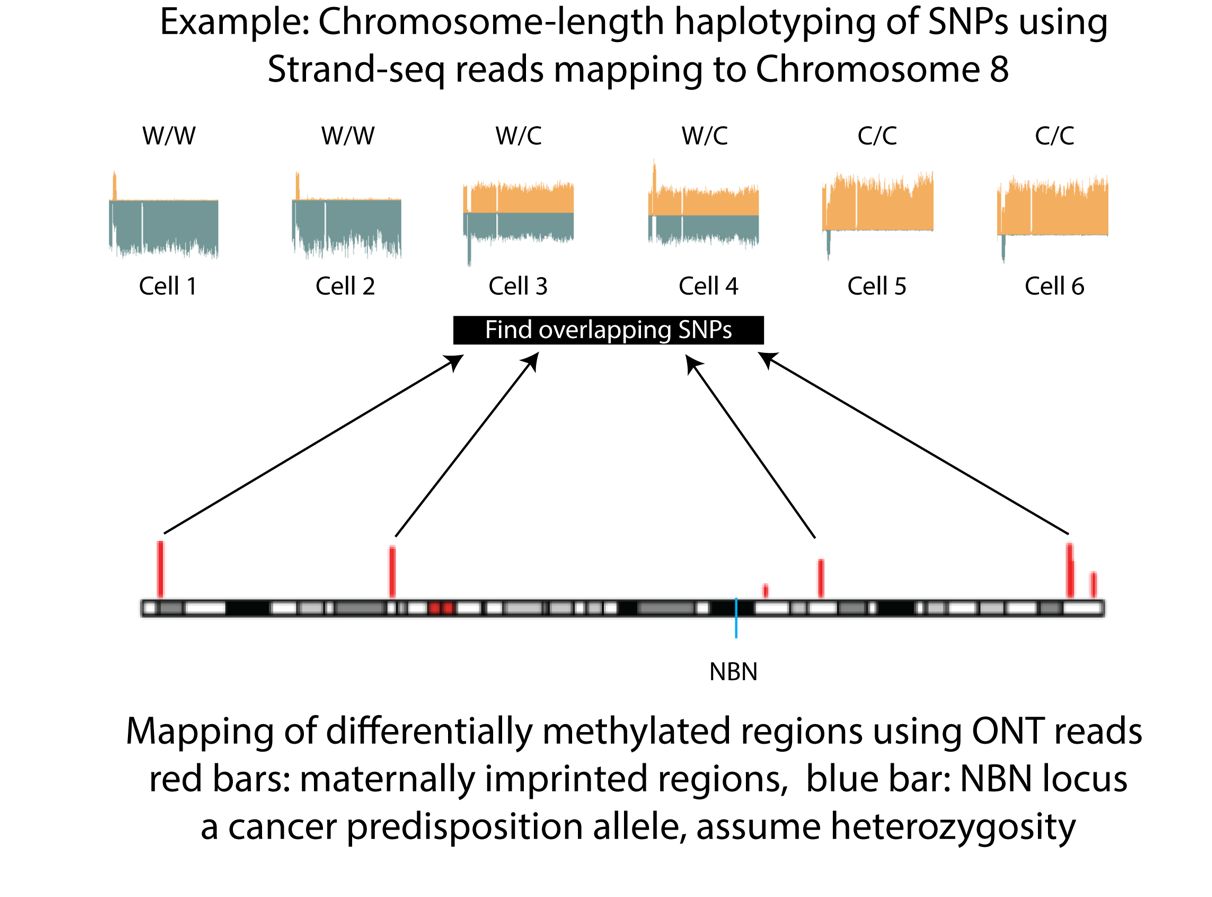
Cells with reads mapping to opposite strands of the reference genome for a particular chromosome occur at a frequency of around 50%. Such cells (3 and 4 in figure) can be used to assign the parent-of-origin haplotype for that chromosome using parental DNA methylation imprints obtained using long read sequencing (1).
References:
- Akbari, V., Hanlon, V.C.T., O’Neill, K., Lefebvre, L., Schrader, K.A., Lansdorp, P.M. and Jones, S.J.M. (2022) Parent-of-origin detection and chromosome-scale haplotyping using long-read DNA methylation sequencing and Strand-seq. Cell Genomics, 100233.
Return to Dr. Lansdorp's profile:

Professor, Medical Genetics, University of British Columbia (UBC)
Distinguished Scientist, Terry Fox Laboratory, BC Cancer
BC Cancer Foundation is the fundraising partner of BC Cancer, which includes BC Cancer Research. Together with our donors, we are changing cancer outcomes for British Columbians by funding innovative research and personalized treatment and care.