Mission and Goals
The Dedhar lab carries out research in the broad area of the Tumour Microenvironment.
Specifically, the goals are to understand how tumour cells communicate with the extracellular microenvironment: matrix proteins, growth factors and other cell types, and to understand, at the molecular and cellular level, how these interactions promote tumour growth and metastasis.
The goals are to identify key signaling pathways and proteins that promote tumour progression and to develop novel therapeutic approaches to target these pathways and proteins to suppress tumour growth and metastasis.
Major Research Areas
- Cell-Extracellular Matrix interactions
- Coordination of Integrin and Growth factor Signal Transduction
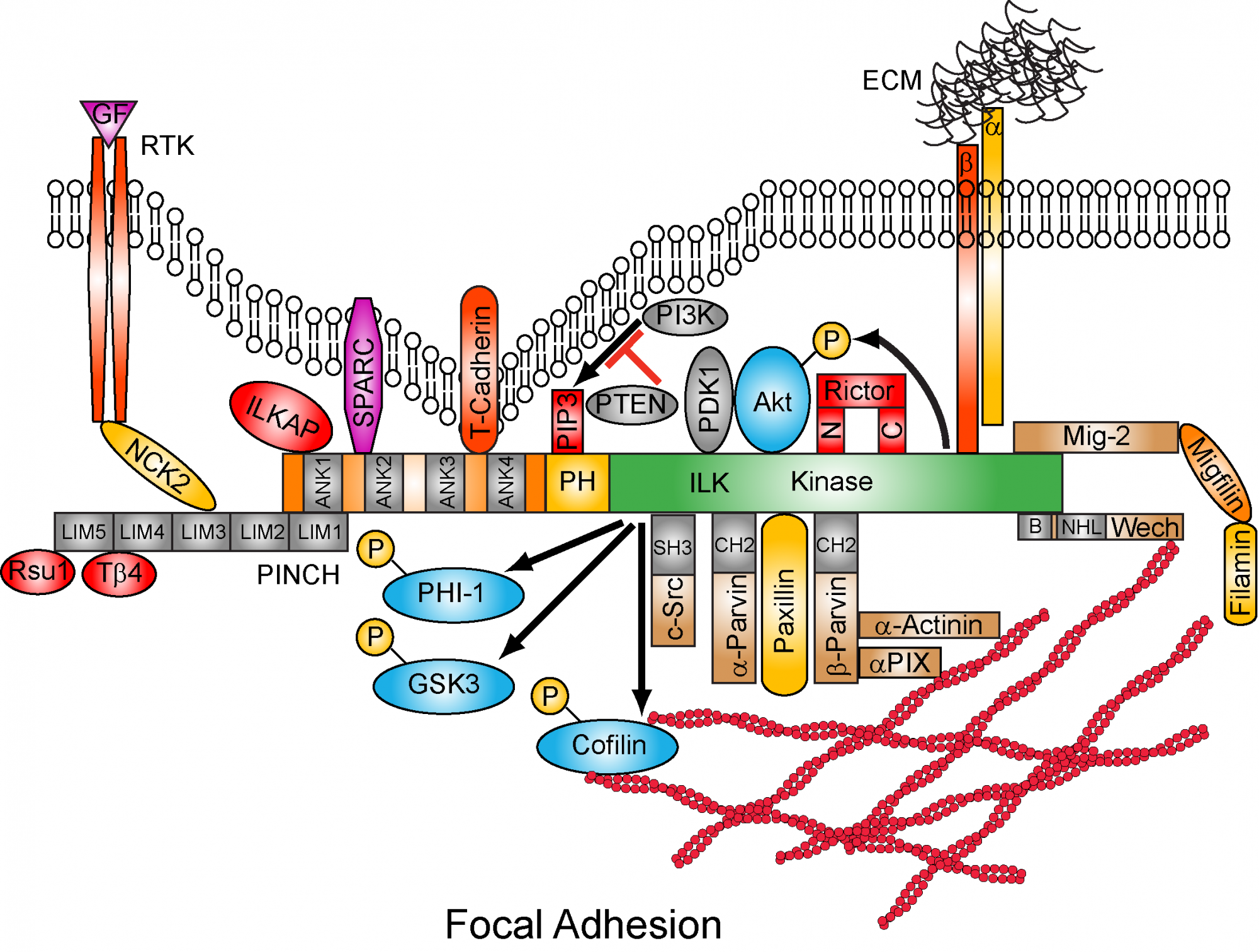
- Physiological and Pathological roles of Integrin-Linked Kinase (ILK)
- Mitotic Spindle Organization and Centrosome Clustering
- Epithelial to Mesenchymal Transition (EMT) and Wnt Signaling
- Models of Metastasis
- Genes Regulating Metastatic Potential
- Hypoxia and Carbonic Anhydrases IX and XII as Therapeutic Targets
- Cancer cell Metabolism and Tumor Microenvironment
Key discoveries have been the identification of Integrin-mediated signaling pathways leading to regulation of cell proliferation, survival and migration. The discovery of Integrin-Linked Kinase (ILK) in 1996 in the Dedhar laboratory has demonstrated how integrin receptors on different types of cells coordinate signals to regulate cellular homeostasis. ILK is required for embryonic development, for the formation of blood vessels and for cardiac function. It is also essential for the regulation of the proper development of neurons as well as the function of other cell types such as chondrocytes, smooth muscle cells, skin cells and hair follicle cells.
ILK was found to induce oncogenic transformation of epithelial cells and subsequently has been shown to be expressed at abnormally high levels in many types of human cancers. In many cases the amount of ILK protein in cells predicts for long term patient survival, i.e. the higher the amount, the worse the prognosis, suggesting that ILK contributes to the disease process and to therapeutic resistance.
ILK activation promotes the survival and growth of tumor cells and also promotes the HIF-1 and VEGF mediated angiogenesis
Recent work using sophisticated Proteomics technology has revealed that ILK localizes to the centrosome, in addition to focal adhesions where integrins are present. In the centrosome ILK controls the assembly of the mitotic spindle during cell division. It also regulates the clustering of supernumerary centrosomes that exist in many cancer cells. The clustering of centrosomes in cancer cells allows them to survive and divide normally. Blocking the function of ILK in centrosomes in such cancer cells results in centrosome “declustering” and cell death. Therefore blocking ILK can induce the death of cancer cells but not of normal cells which do not require centrosome clustering.
ILK is a Cancer Therapeutic Target.
Inhibition of ILK activity suppresses tumor growth and progression to metastasis, and compounds that inhibit ILK activity are under development. Inhibitors developed so far appear to selectively kill tumor cells relative to “normal” counterparts. Collaborative research has shown that ILK inhibitors could be used therapeutically for breast cancer, prostate cancer, pancreatic cancer, colon cancer, non small cell lung cancer, malignant melanoma, glioblastoma, acute myelogenous leukemia ( AML), chronic myelogenous leukemia (CML)
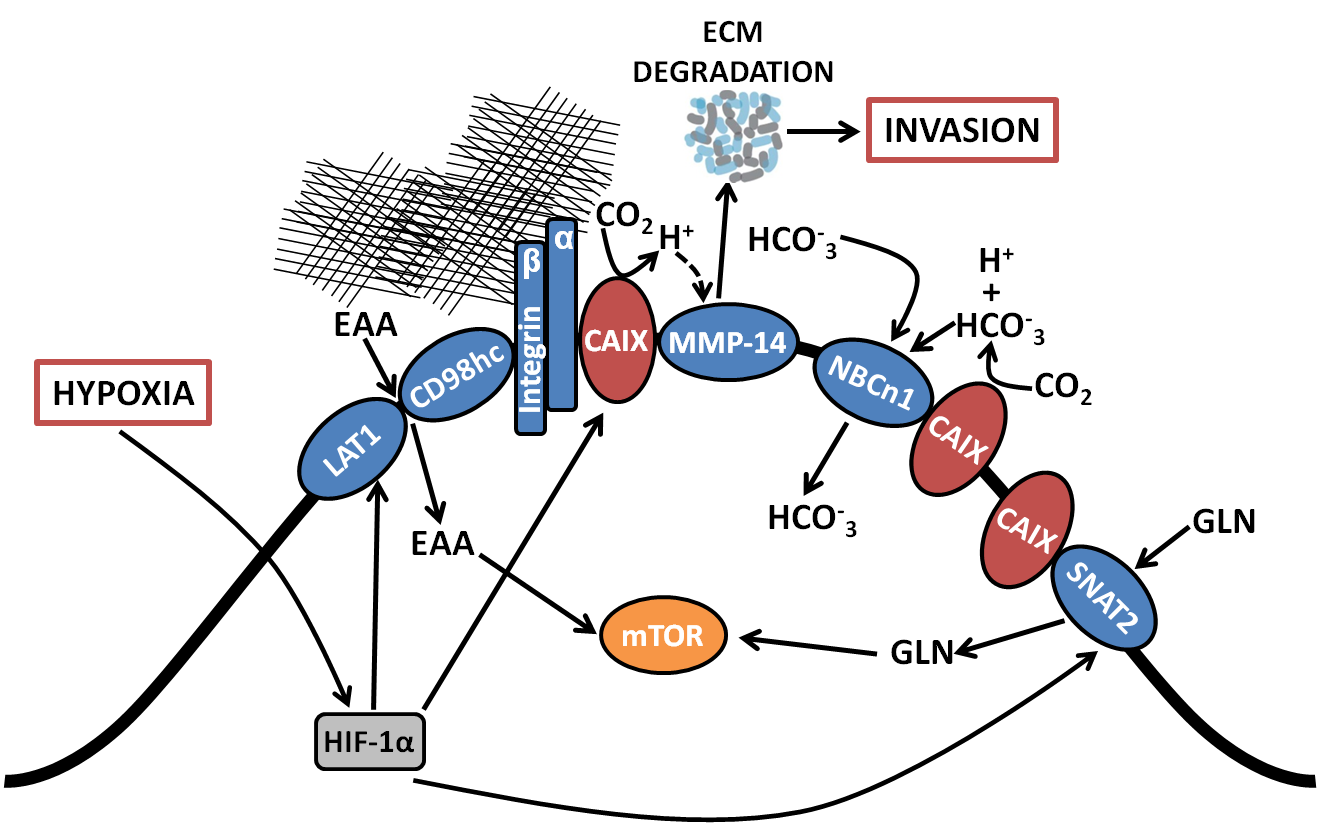
Models of Cancer Metastasis, Tumour Hypoxia and Carbonic Anhydrases IX and XII
Patient mortality results from the formation of metastases in distant (vital) organs. The metastatic potential of primary tumors is variable and likely depends of the biology of the tumor. We have established mouse models to study metastasis. Our initial work with breast cancer models of metastasis has identified a gene signature that is unique to metastatic tumors. Many of the genes are induced by “hypoxia” and function to protect the cells and promote survival in a hypoxic environment. Two of the genes CAIX and CAXII regulate the pH of the cells in hypoxia, and we have discovered that blocking the function of CAIX results in the death of the metastatic cells in a hypoxic environment, and in tumor regression as well as formation of metastasis.
Examination of the expression of CAIX in about 4000 breast cancer samples demonstrated that CAIX expression predicts for poor patient prognosis as well as for the formation of distant metastasis. Current work is focused on determining how CAIX controls the survival of hypoxic tumor cells and why it is needed for the formation of metastases. We are also developing compounds that could be used therapeutically to block CAIX function and also to image CAIX and identify hypoxic and potentially metastatic tumors.
BioID proteomic analysis has revealed that CAIX forms a large protein complex with many metabolic transporters, integrins and matrix modeling enzyme, MMP-14. The CAIX/MMP-14 complex localizes to invadopodia and promotes collagen degradation, invasion and metastasis.
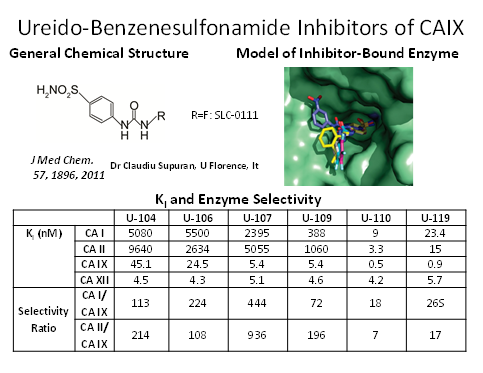
We have developed a highly selective small molecule inhibitor (SLC-0111) of CAIX/XII, and demonstrated that it can inhibit tumor growth and metastasis of breast cancer, pancreatic cancer and glioblastoma. Recent work has shown that the CAIX inhibitor can be combined with immune check-point inhibitor antibodies to improve survival in pre-clinical models of melanoma and breast cancer. The compound SLC-0111 has been evaluated for safety and pharmacokinetics in a Phase 1 clinical trial, and is currently being evaluated in a Phase 1b, SLC-0111/gemcitabine combination clinical trials in CAIX-positive pancreatic cancer patients.
Evaluation of “Anastasis”: “recovering from the brink of death” as a mechanism of resistance of cancer cells to cancer chemotherapy drugs.
Anastasis is a process that has been identified in cells undergoing apoptosis induced through various stresses. Cells that are already committed to cell death through the activation of the “executioner” caspases have been shown to recover and survive through as yet poorly understood mechanisms.
The Dedhar lab is carrying our research into identifying the mechanisms whereby cancer cells exposed to clinically used cancer chemotherapy drugs recover from the brink of death, i.e. undergo anastasis.
This project is in its infancy, but preliminary results point to novel cellular mechanisms including the expression of a variant splice form of Caspase 3, accumulation of stress granules containing messenger RNAs poised for translation, and epithelial to Mesenchymal Transition ( EMT). The goals of the project are to identify central players in the anastasis process and to target them to overcome anastasis and drug resistance.



